 Por Francisco Siredey
Por Francisco SiredeyLucas: En busca de la cura de los dos millones de dólares
Una pareja iquiqueña necesita adquirir el medicamento más caro del mundo para poder salvarle la vida a Lucas, su hijo. Deben hacerlo antes de que el niño cumpla dos años. En su carrera contra una extraña enfermedad genética descubrieron que cientos de familias chilenas viven lo mismo.

Recibieron el sobre el 24 de julio. Marcela Márquez y su marido, Álvaro Leiva, no se decidían a abrirlo por temor a los resultados. El duro diagnóstico que la Dra. Consuelo Gayoso les había entregado cuatro días antes solo sería real si la información contenida en esa hoja de papel así lo estableciera; ambos tenían la esperanza de que todo fuera un error. Cuando finalmente se envalentonaron, el estudio genético les confirmó que la neuróloga tenía razón: su hijo Lucas, de solo tres meses, tenía Atrofia Muscular Espinal (AME), una extraña enfermedad que afecta a uno de cada 10.000 niños en el mundo.
Hasta pocas semanas antes del diagnóstico, ninguno había escuchado hablar de esta patología, pese a que los dos son profesionales de la salud: Marcela Márquez es matrona y Álvaro Leiva es dermatólogo. Ambos se conocían desde 2008, como compañeros de trabajo del Hospital Regional de Iquique. Al poco tiempo, iniciaron una relación de pareja. Además de seguir profesiones similares, también tenían en común la afición por el running, que los llevaba a participar en carreras extremas por todo Chile.
La inquietud por ser padres solo apareció un par de años atrás, después de una beca de tres meses que Álvaro Leiva cursó en Sao Paulo. Su deseo se materializó a mediados de 2019. A las pocas semanas de embarazo, supieron que esperaban mellizos. Lucas y Victoria Leiva nacieron el 19 de abril de este año, en el Hospital San Juan de Dios de La Serena, lejos del hogar de sus padres en Iquique. Como el parto sería prematuro y en Tarapacá las camas críticas se estaban llenando rápidamente con pacientes Covid-19, la pareja decidió trasladarse a la Región de Coquimbo, una zona que, por entonces, estaba menos afectada por la pandemia. Decidieron quedarse ahí hasta que los contagios decayeran.
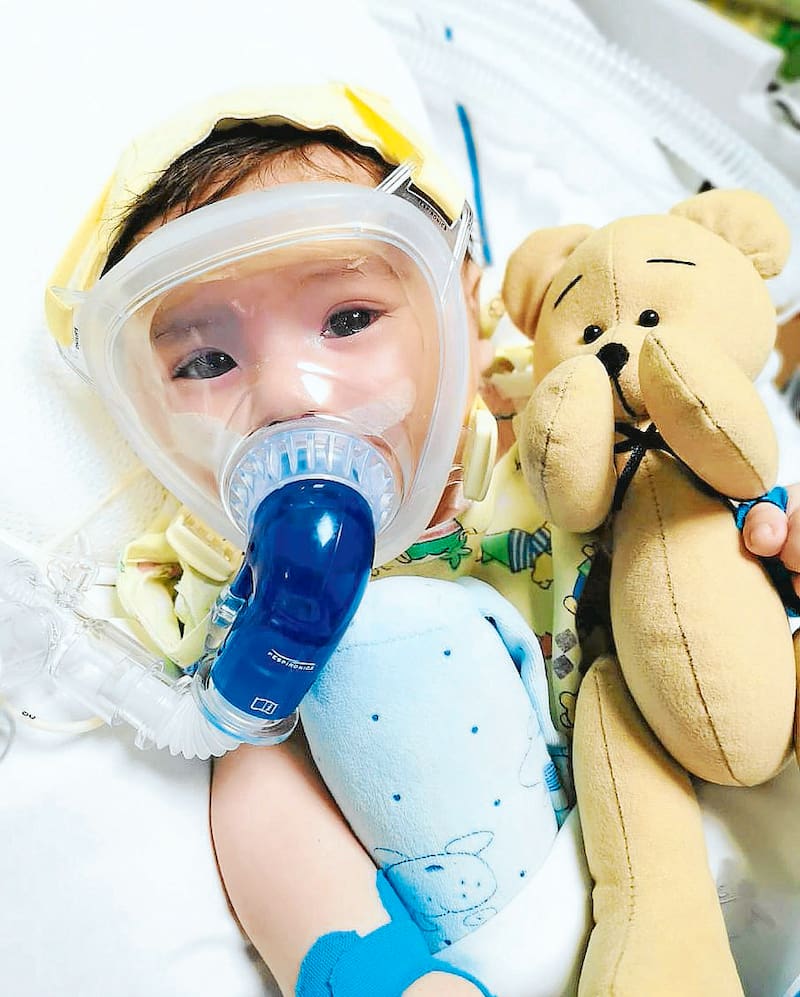
A fines de junio empezaron a notar que Lucas no era capaz de mantener la cabeza erguida y que estaba teniendo problemas para tomar la leche materna. Eran los primeros síntomas de la AME tipo 1, una enfermedad genética que le impide al cuerpo producir la proteína SMN-1, que es la que permite la sobrevivencia de las neuronas que controlan las funciones motoras. Este mal se manifiesta muy tempranamente y va deteriorando los músculos de manera progresiva, hasta que los pacientes ya no pueden moverse, comer o respirar por su cuenta. La gravedad de estos efectos reduce la esperanza de vida al mínimo: apenas dos años.
A nivel mundial, la AME es la primera causa genética de mortalidad infantil.
"Saber que un hijo se te puede morir de los dos años en adelante, no sé si existe más dolor que eso. Ahora puedo hablar sin llorar, pero hasta hace poco me quebraba”, dice Marcela Márquez. “Eran hijos tan deseados. Uno espera que ellos te entierren, que corran juntos… Pensar que no los vas a tener contigo es desgarrador”.
La hermana de Lucas no había mostrado síntomas, pero tenía un 25% de posibilidades de sufrir la misma enfermedad. Los padres volvieron a pasar por la agonía de esperar los resultados, hasta que estos indicaron que Victoria no tenía AME y solo portaba el gen recesivo.
Como la isapre Consalud contemplaba como prestadora preferencial para AME a la Clínica Dávila, la familia se trasladó a Santiago. Lucas permanece hospitalizado ahí desde el 7 de agosto. Su mamá lo acompaña durante la semana, su melliza permanece bajo el cuidado de sus abuelos maternos y su padre viaja al norte cada semana para trabajar. Necesitan entradas de dinero constantes para cubrir el tratamiento.
“El buque no se puede parar”, comenta Álvaro Leiva. “Es una agonía estar pensando en ellos todo el día y estar solo. No puedes juntarte con nadie. Eso castiga más. Vivo con mascarilla todo el día en las consultas, contando los días para volver”.
Si la familia ya estaba abatida con la situación, quedaron casi paralizados al enterarse de que existe una cura para la enfermedad de Lucas, pero que su precio bordea los dos millones de dólares -cerca de 1.600 millones de pesos- y que debe ser administrada antes de los dos años para poder salvarle la vida al paciente.
***
Marcela Márquez todavía no aceptaba el diagnóstico de Lucas cuando recibió el llamado de Paulina González, la presidenta de Fame, la Corporación Familias Atrofia Muscular Espinal de Chile. No estaba con demasiado ánimo para conversar, pero en ese diálogo se enteró de que en Chile hay al menos 300 familias en las mismas circunstancias.
La agrupación se remonta al año 2010, cuando un grupo de familiares de pacientes se encontró en la Teletón, donde muchos niños y jóvenes realizaban terapias de rehabilitación. Como entonces no existía ningún medicamento, se trataba de pacientes con AME tipo 2 y 3, cuadros menos severos que se manifiestan después de los dos años y que representan el 40% del total de los casos. A través de una acción coordinada, los fundadores lentamente fueron logrando triunfos judiciales para que sus seguros de salud, tanto Fonasa como isapres, respondieran a los enormes gastos del tratamiento. Además de las hospitalizaciones en centros asistenciales o domiciliarias, muchos pacientes requerían ventilación mecánica, sondas de alimentación y diversas máquinas para sobrevivir. También necesitaban kinesiólogos, terapeutas ocupacionales, fonoaudiólogos y acceso a ejercicios en piscina.
Recién hace dos años apareció el primer medicamento para frenar el avance del AME. Se llama Spinraza, del laboratorio Biogen. El costo de cada dosis es altísimo, cerca de $ 90 millones, y solo durante el primer año se deben inyectar seis por vía intratecal. Hasta la fecha, según Paulina González, alrededor de 30 familias que se atienden en el sistema público han ganado su acceso a este fármaco en tribunales, demandando al Estado. Otras han conseguido que sus isapres cubran el tratamiento, aunque pagando deducibles que no dejan de ser millonarios.
También hay algunos casos en que los mismos tribunales han rechazado los recursos, como le ocurrió recientemente a una pareja con dos hijos AME 2, cuya acción legal fue desestimada por la Corte Suprema, luego de un fallo favorable de la Corte de Apelaciones.
“Pedimos que el Estado nos incluya en una ley. Siempre hemos quedado fuera de la Ley Ricarte Soto”, comenta Paulina González, quien además de presidenta es madre de una niña de nueve años con AME tipo 2. “Estamos reiniciando conversaciones con el Minsal, para que nos puedan incluir. No es nuestra intención seguir demandando al Estado, que está gastando mucho pagando esas demandas. Se puede acceder a mejores precios”.
Al Spinraza pronto se le unirá Risdiplam, de Roche, que también frena el deterioro neuronal y está recomendado especialmente para pacientes de AME tipo 2 y 3, que tienen una esperanza de vida que bordea los 20 años. Sin embargo, ninguna de estas dos drogas es efectiva a largo plazo entre los menores de seis meses que sufren de AME tipo 1.
Para los casos más graves, como el de Lucas, la única alternativa para sobrevivir es Zolgensma -o AVXS-101-. En 2019, el laboratorio suizo Novartis liberó esta terapia de aplicación única, que introduce al organismo un adenovirus que simula el gen faltante. Este vector estimula la producción de la proteína SMN-1 que permite la sobrevivencia de las neuronas motoras. De esta forma, actuaría sobre el origen de la enfermedad y podría permitirles a los pacientes un desarrollo muscular normal. Para que el tratamiento sea efectivo, sin embargo, se necesita de un diagnóstico temprano y una acción rápida, pues las neuronas que van muriendo no se recuperan.
Zolgensma tiene un inconveniente más importante. A un precio de dos millones de dólares, es el medicamento más caro del mundo. Pese a que ha sido aprobado en EE.UU., la Unión Europea, Japón y Brasil, aún no son demasiadas las aseguradoras dispuestas a cubrirlo.
“Efectivamente, es muy costoso. Fabricarla es muy complicado. Requiere alta tecnología y eso genera un costo para la compañía. Es un medicamento customizado. Se envía la información de cada paciente a Chicago y de acuerdo con eso se hacen los viales. Pero si lo pones junto a Spinraza, que hay que administrarlo de por vida, se paga en tres o cuatro años”, explica Lucelia Tavares, gerenta médica para AME de Novartis.

Hasta el momento, en Chile solo hay dos pacientes que han sido tratados con Zolgensma: Mariana Bozo fue parte de los ensayos clínicos en Boston, y Luciana Abarca, literalmente, se ganó la lotería. Debido al exorbitante costo del medicamento, Novartis organizó un sorteo de 100 dosis anuales entre pacientes AME de todos los países donde Zolgensma todavía no está aprobado. “Lucianita”, de familia talquina, fue la primera ganadora chilena de la “tómbola” y ya recibió con éxito la infusión en la Clínica Las Condes.
Aunque los sorteos de la farmacéutica se efectúan cada dos semanas, los padres de Lucas no pueden inscribirlo. Uno de los criterios para participar es que los pacientes no hayan recibido ningún otro tratamiento contra la AME. En su caso, Marcela Márquez y Álvaro Leiva optaron por administrarle Spinraza a su hijo con el objetivo de frenar el avance de la enfermedad y ganar tiempo. Su isapre se comprometió a cubrirles las primeras seis dosis contra el pago de un deducible de cinco millones. Ninguno de los dos padres entiende que esa decisión los descalifique de la lotería por la cura.
“Es una desesperación tremenda para un padre saber que existe una cura y no poder acceder a ella. No se lo doy a ninguna familia. Cuando supimos que existía lo vimos muy lejos. Ni siquiera pensamos en eso”, dice Marcela Márquez.
La madre de Lucas cree que se debe al interés de Novartis por verificar que los resultados posteriores sean inequívocamente atribuibles a Zolgensma y no a otra droga como Spinraza.
Desde el laboratorio suizo aseguran que el motivo es otro:
"Si el paciente tiene acceso a otra terapia en el país, no puede participar, porque le quita la opción a quienes no tienen ninguna. La idea es darles acceso a quienes no tienen alternativas”, dice Tavares.

***
En Iquique, los amigos y compañeros de trabajo de Marcela Márquez y Álvaro Leiva han organizado una robusta campaña para reunir fondos para Lucas. Fue Carlos Reyes, un amigo dermatólogo del padre, quien convenció a ambos de que al menos había que intentarlo. Las dificultades de la pandemia han impedido actividades presenciales, pero virtualmente se están rifando premios como un auto, una cirugía estética y un tratamiento dental. La meta son los 1.600 millones que cuesta una dosis de Zolgensma; hasta el momento, han reunido 186 millones, poco más del 10%. Para poder atraer contribuciones más grandes, están tramitando la personalidad jurídica de una fundación que les permita recibir donaciones de empresas.
“Yo soy sureño, de Contulmo, pero me siento iquiqueño ahora”, dice Álvaro Leiva, agradecido de la ciudad donde ha vivido los últimos 12 años.
Actualmente, Lucas se encuentra estable en Santiago, con apoyo respiratorio. Reacciona a diversos estímulos, se ríe y llora. Ya ha recibido tres dosis de Spinraza y en los próximos días recibirá la cuarta. El deseo de ambos papás es que Lucas pueda recibir el medicamento antes de su primer cumpleaños, para así evitar que sufra algún daño neuronal irreversible. Lógicamente, tienen serias dudas de poder alcanzar la cifra por sus medios, por lo que también apuestan a las gestiones que Fame está realizando con el Minsal.
Este martes, la directiva de la corporación sostendrá una cita de coordinación con el ministro Enrique Paris, quien comenta que conversó con el padre de Lucas y que la primera dama Cecilia Morel también está al tanto de su caso. El secretario de Estado considera que en vez de incluir esta patología en la Ley Ricarte Soto, se podría usar la nueva ley de Cenabast para negociar una cantidad de dosis con Novartis y venderlas sin intermediarios.
“Estamos en contacto con los especialistas y creo que podemos importar una cantidad importante a bajo precio y venderlos a un precio justo”, dice Paris.
En la filial chilena de Novartis esperan una conversación con el gobierno para buscar alguna fórmula de subsidiar el medicamento entre ambas partes. Antes de eso, advierten, es necesario que el Instituto de Salud Pública (ISP) le dé una aprobación expedita a Zolgensma, cuya documentación será presentada en los próximos meses.
A la espera de alguna novedad en el frente institucional, Marcela Márquez y Álvaro Leiva esperan que Lucas pueda pasar a un régimen de hospitalización domiciliaria en las próximas semanas. Esto les permitiría reunirlo con Victoria, su melliza, y poder descansar un poco tras cinco meses de estrés absoluto. Ambos padres creen que solo han logrado superar este trance gracias a la energía que, pese a todo, ven en su hijo.
“Sus ojos son tan expresivos, que te transmiten cosas. Siento que me dicen ‘mamá, no decaigas’”.
*Para ayudar a la familia de Lucas, se puede depositar cualquier aporte a nombre de Marcela Márquez Luza (RUT: 13.867.497-5) a su cuenta corriente de BancoEstado (No. 1300002125). La notificación se puede hacer a marcelamarquezluza@gmail.com.
COMENTARIOS
Para comentar este artículo debes ser suscriptor.
Lo Último
Lo más leído
La mayoría no entiende el debate por el impuesto a las empresas. El resto lee La Tercera.
50% Plan Digital+$5.150 al mes SUSCRÍBETE













